Following prolonged therapy with afatinib, many independent afatinib-resistant
http://caspasesignaling.com/foll ... fatinib-resistant-c
Right after prolonged treatment method with afatinib, many independent afatinib-resistant clones emerged that persisted and proliferated even within the presence of two ?M afatinib. From these, we established 14 various independent clones that acquired resistance to afatinib. A few of the drug responses with the representative resistant clones are CYP17 Inhibitors shown in Fig. 1B. To keep track of no matter if the acquisition of resistance was irreversible, we cultured all of these clones in standard development medium with no the RTK inhibitor. These clones remained resistant to afatinib soon after incubation in standard development medium for quite a few months , indicating that these cells underwent an irreversible alter, potentially involving chemically stable physical alterations in their genetic space. The signature exon 19 deletion of parental PC9 cells remained unaltered in all of the r . Full surgical resection of tumors or liver transplantation is only probable in a minority of patients; for patients with innovative condition, the prognosis is extremely poor, with an overall median survival of only some months. Response rates to classical chemotherapy are reduced, and also with combination regimens, long lasting remission has remained elusive . Thus, there is a solid need for added therapeutic possibilities.
Lately, rationally created, molecularly targeted medicines have grown to be out there. These agents are built to target growth or survival pathways hyper-activated in cancer cells. Tyrosine kinases are considered to become exceptional molecular oncology targets Dabigatran due to the fact they transduce growth and survival signals and therefore are hyper-activated in many, if not all, human malignancies . The ErbB receptor tyrosine kinase family members, comprising EGFR and ErbB2, -3, and -4, has become of central interest during the development of targeted anticancer strategies. Trastuzumab , a monoclonal antibody against ErbB2, is efficiently being utilized in individuals with ErbB2-overexpressing breast cancer, and overexpression of ErbB2 through gene amplification is really a excellent predictor of favorable response . Several preclinical and clinical research have addressed the efficacy of EGFR-targeting agents, including tyrosine kinase inhibitors , for example gefitinib and erlotinib , likewise as monoclonal anti-EGFR antibodies, like cetuximab, for that treatment of non-small cell lung cancer , head and neck cancer, colon carcinoma, glioblastoma, together with other tumors . Even though NSCLC sufferers with activating mutations inside the kinase domain of EGFR respond favorably to EGFR TKIs, top to their approval for this subset of malignancies, the molecular basis figuring out the response of tumor cells to EGFR-targeting drugs in other settings is only partially understood and it is talked about controversially.
|
 都说免疫不入脑,入了就不得了
免疫治疗以后的疗效反应,当病灶增大时,不一定是进展,还有一种可能,叫做CR,这个现
都说免疫不入脑,入了就不得了
免疫治疗以后的疗效反应,当病灶增大时,不一定是进展,还有一种可能,叫做CR,这个现
 晚期肺癌经过4次换药,如今母亲已经4
作者:pears
“万事开头难”这句话在母亲的抗癌路上体现得淋漓尽致,最初确诊肺癌后半
晚期肺癌经过4次换药,如今母亲已经4
作者:pears
“万事开头难”这句话在母亲的抗癌路上体现得淋漓尽致,最初确诊肺癌后半
 既要,又要,还要,什么药品可以治疗
作者:seacat
EGFR突变可以分为常见突变和罕见突变。常见突变就是EGFR外显子19del突变
既要,又要,还要,什么药品可以治疗
作者:seacat
EGFR突变可以分为常见突变和罕见突变。常见突变就是EGFR外显子19del突变
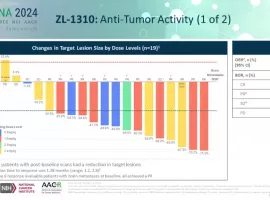 小细胞肺癌新希望?标准治疗耐药后OR
作者:Keenman
如果以药物规模化获批进入临床的时间来划分的话,进入21世纪以来,肺癌
小细胞肺癌新希望?标准治疗耐药后OR
作者:Keenman
如果以药物规模化获批进入临床的时间来划分的话,进入21世纪以来,肺癌
 母亲的基因检测和PD-L1报告出来了,
看了基因检测报告,似乎没有合适的靶向药,母亲确诊肺腺癌晚期,求助大家帮忙分析一下
母亲的基因检测和PD-L1报告出来了,
看了基因检测报告,似乎没有合适的靶向药,母亲确诊肺腺癌晚期,求助大家帮忙分析一下





 显身卡
显身卡

